Graphical models for variant aggregation
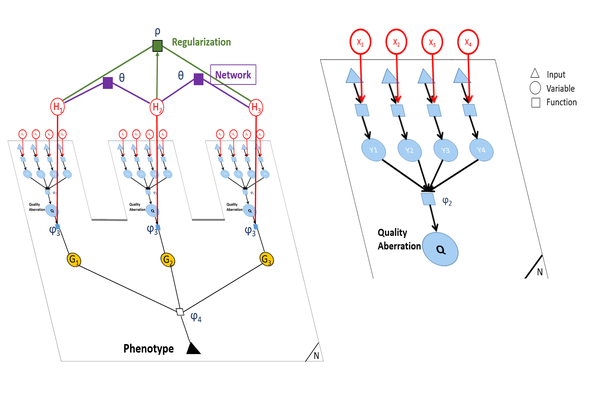
Discovering genetic mechanisms driving complex diseases is a hard problem. Existing methods often lack power to identify the set of responsible genes. Protein-protein interaction networks have been shown to boost power when detecting gene-disease associations. We introduce a Bayesian framework, Conflux, to find disease associated genes from exome sequencing data using networks as a prior. There are two main advantages to using networks within a probabilistic graphical model. First, networks are noisy and incomplete, a substantial impediment to gene discovery. Incorporating networks into the structure of a probabilistic models for gene inference has less impact on the solution than relying on the noisy network structure directly. Second, using a Bayesian framework we can keep track of the uncertainty of each gene being associated with the phenotype rather than returning a fixed list of genes. We first show that using networks clearly improves gene detection compared to individual gene testing. We then show consistently improved performance of Conflux compared to the state-of-the-art diffusion network-based method Hotnet2 and a variety of other network and variant aggregation methods, using randomly generated and literature-reported gene sets. We test Hotnet2 and Conflux on several network configurations to reveal biases and patterns of false positives and false negatives in each case. Our experiments show that our novel Bayesian framework Conflux incorporates many of the advantages of the current state-of-the-art methods, while offering more flexibility and improved power in many gene-disease association scenarios.